摘要
根据基因表达的总体差异,乳腺癌被分为预后和治疗反应不同的亚型。管腔乳腺癌的基因表达和增殖是由雌激素受体α驱动的,靶向该转录因子是治疗这一亚型的最有效的方法。相比之下,目前还不清楚是哪些转录因子驱动了基底样三阴性乳腺癌的基因表达,也没有批准有针对性的疗法来治疗这种侵袭性亚型。在这项研究中,我们综合分析了大量的乳腺癌细胞系和患者肿瘤组织的DNA甲基化、染色质可及性、转录因子结合位点和基因表达等基因组学数据,以鉴定与基底样基因表达程序有关的转录因子。我们发现,糖皮质激素受体(GR)和STAT3结合到相同的基因组调控区,这些区域在基底样乳腺癌中是特异开放且非甲基化的。这些转录因子共同调节了数百个基底样基因的表达,且这些基因与预后不良有关。两种转录因子的小分子抑制剂联合治疗导致细胞系和患者衍生的器官模型中的细胞生长协同减少。本研究证实了GR和STAT3协同调节基底样三阴性乳腺癌基因表达的程序,为合理联合STAT3和GR抑制剂改善基底样三阴性乳腺癌的治疗提供了依据。
Introduction
根据总体基因表达差异,乳腺癌可分为预后和治疗反应不同的亚型。这些表达差异反映了肿瘤从中产生的正常细胞的转录因子(TF)、表观遗传状态和基因调控 络,以及它们在肿瘤发生过程中积累的变化。
正常乳腺细胞通过激活TF雌激素受体(ER)α和ER下游靶基因,对雌激素作出反应而增殖。表达ER的乳腺癌最常分为管腔亚型。这些管腔ER肿瘤用激素疗法治疗,以阻止ER激活其增殖转录程序。尽管这些肿瘤通常有野生型内质 ,但这是一种非常有效的靶向治疗,因为它抑制了这类细胞中基因表达和增殖的主要调节因子。
相比之下,15%至20%的乳腺癌患者被诊断为三阴性乳腺癌(TNBC),三阴性乳腺癌的定义是指雌激素受体、孕激素受体及人表皮生长因子受体2均阴性的乳腺癌患者。目前还没有FDA批准的靶向治疗TNBC。即使经过手术、放疗和细胞毒化疗的积极治疗,TNBC患者在3年内的复发率也很高,疾病死亡率也很高(38%)。对于TNBC来说,迫切需要替代的治疗策略。TNBC是一种分子异质性疾病,已有大量研究发现TNBC肿瘤亚群具有明显的分子特征。这些亚型包括具有与上皮向间充质转化相关的特征的肿瘤,通常被称为claudin-low或间质亚型,以及由雄激素受体(AR)而不是ER,现在通常被称为管腔雄激素受体亚型(LAR),它对抗雄激素治疗有反应。TNBC最广泛的特征是基底样亚型,它区别于ER驱动的管腔乳腺癌。根据PAM50分型,约70%~80%的TNBC肿瘤为基底样型乳腺癌,基底样型和腔型乳腺癌之间存在数千个基因的差异表达,基底样型乳腺癌中表达上调的基因反映了TNBC生长快、预后差的特点。目前还不清楚哪些TF负责调节基底样亚型中的增殖转录程序。本研究的目的是确定在基底样乳腺癌中调节临床相关基因调控程序的因子,并确定抑制这些因子是否可以提供新的治疗策略来改善基底样癌患者的临床结果。
许多TF在基底细胞样乳腺癌中的作用已被研究。许多这样的转录因子调节基底样乳腺癌中的重要通路,而且这些转录因子经常相互调节彼此的表达。其中几种TF可以被小分子药物抑制,是很有希望的治疗靶点。目前尚不清楚的是,哪些TF负责驱动定义基底样亚型的基因表达特征,哪些TF位于主调控因子下游。
这项研究利用DNA甲基化、染色质可及性、TF结合、基因表达和细胞生长的综合分析,在大量乳腺癌细胞系和患者肿瘤中识别驱动基底样基因表达程序的TF。我们试图找出与腔性乳腺癌相比,基底样染色质中特别未甲基化且可获得染色质的调控区域。我们确定了其基序和结合位点在这些碱基特异调控区域高度富集的TF。然后,我们调查了这些TF是否在基底样癌中比管腔性乳腺癌更高表达,这些TF是否调控基底样基因表达程序中的基因,以及这些TF调控的基因是否与患者的预后相关。这种全基因组的方法使我们发现,两个TF[STAT3和糖皮质激素受体(GR)]之间的合作,而不是单一的“主调节器”,推动了基底样基因表达程序中数百个基因的表达。这种TF相互作用为基底样细胞提供了弹性生长能力。此外,在基底样细胞系和患者衍生的器官模型中,同时抑制两种TF导致细胞生长协同减少,这为基底样乳腺癌提供了一种新的治疗策略。
材料和方法
细胞系
本研究使用了以下基底样乳腺癌细胞系:HCC1569、HCC1954、BT-20、HCC1143、HCC1187、HCC1599、HCC1937、HCC70、MDA-MB-468、2-LMP、BT-549、HCC38、MDAMB-157、MDA-MB-231、MDA-MB-436、SUM102、SUM149和SUM159。
本研究以DY36T2、ZR-75-30、MDA-MB-453、SK-BR-3、BT-474、MDAMB-361、MCF-7、MDA-MB-134、T-47D和ZR-75-1为研究对象。
传代超过3个月的细胞系每6个月使用由犹他州大学DNA测序核心设施提供的短串联重复测试服务进行验证。
简并代表性亚硫酸氢盐测序技术(RRBS)
使用前面描述的方法在对细胞系进行简并代表性亚硫酸氢盐测序(RRBS)和CG甲基化的初步分析。在至少80%的细胞系中具有至少10倍覆盖率的CG位点被用于后续分析。为了鉴定在基底样细胞系和管腔细胞系之间差异甲基化的CG,在R软件包版本3.5.0中使用lm函数进行线性回归。使用R中的p值校正函数和Benjamini-Hochberg得方法校正P值以进行多重假设检验。校正后的P值小于0.05的CG使用pheatmap包版本1.0.10在R中可视化。RRBS数据可通过NCBI Gene Expression Omnibus(GEO)登录 GSE152202公开获得。
ENCODE TF结合位点的富集
从UCSC基因组浏览器下载名为wgEncodeRegTfbsClusteredV2.bed.gz的文件,该文件包含由ENCODE项目在各种细胞系中对149个TF进行的染色质免疫沉淀测序(ChIP-seq)实验中的TF结合位点。使用Bedtools交集来确定RRBS数据集中的哪些CG位置与文件中每个TF的结合位点位置重叠。与基底样CGs和管腔特异性CGs的重叠数归一化为RRBS中所有CGs的重叠数,以计算折叠富集。
肿瘤基因组图谱RNA测序及ATAC-seq分析
癌症基因组图谱(TCGA)乳腺癌样本的RNA测序(RNA-seq)数据文件是从UCSC Xena浏览器下载的。这些文件包含日志数据,因此在R版本3.5.0中将它们转换为可用数据。利用TCGA提供的PAM50亚型分类,DESeq2版本1.20.0用于识别基底样瘤和管腔瘤之间差异表达的基因,并可通过UCSC Xena浏览器下载。使用校正后的P值
转座酶可及染色质高通量测序分析(ATAC-seq)来自TCGA乳腺肿瘤的数据以counts文件和bigwig文件的形式从Corces及其同事最近出版的《补充资料》中下载。利用TCGA提供的PAM50亚型分类,DESeq2版本1.20.0用于识别基底样瘤和管腔瘤之间可区别访问的峰,并可通过UCSC Xena浏览器下载。Pheatmap软件包版本1.0.10用于可视化使用校正后的P值
为了分析基底特异性GR和STAT3共享结合位点上的TCGA乳腺肿瘤的ATAC-seq信 ,使用UCSC hgLiftOver将GR和STAT3共享位点的文件转换为hg38基因组构建,以与TCGA ATAC-seq比对兼容。Deeptools版本3.1.0 computeMatrix 用于为每个样本生成GR和STAT3共享站点的数据矩阵。使用R版本3.5.0计算每个样本跨区域的平均可及性。我们使用Mann-Whitney检验来确定基底样肿瘤和腔内肿瘤间的平均归一化ATAC-seq跨区域读取深度是否有显著差异。使用GraphPad Prism 7.0c绘制基底样和管腔样本的平均归一化深度的平均值和标准差。使用VIOPLOT_0.3.2创建GR和STAT3共享部位基底样瘤和管腔肿瘤的平均bigwig读数的violin plots,使用默认参数和等面积=T,h=0.04,Wex=0.9。
ChIP-seq
对4株基底样细胞(SUM159、MDA-MB-231、HCC1937、HCC70)和4株管腔细胞(MDA-MB-361、BT-474、MDA-MB-453、MCF-7)进行GR和STAT3的ChIP-seq。ChIP-seq实验中,蛋白质- DNA复合物通过1%甲醛在室温下共价交联10分钟。用0.125 mol/L甘氨酸孵育5分钟,以猝灭交联反应。用PBS (pH 7.4;Lonza)。用含蛋白酶抑制剂(Roche)的Farnham Lysis Buffer (pH 8.05 的5mmol/L PIPES, 85 mmol/L KCl, 0.5% NP40)裂解细胞。细胞裂解液于4℃,2000 rpm离心5min。上清液粗提的细胞核于-80℃保存。使用GR抗体(sc-1003,Santa Cruz Biotechnology)和STAT3抗体(sc-482,Santa Cruz Biotechnology)进行ChIP-seq。这项研究中使用的ChIP-seq协议的完整版本可在ENCODE项目 站上获得:https://www.encodeproject.org/documents/df9dd0ec-c1cf4391-a745-a933ab1af7a7/@@download/attachment/Myers_Lab_ChIPseq_Protocol_v042211.pdf
STAT3和GR 的ChIP-seq数据已保存在NCBI GEO登录 GSE85579和GSE152203中。将ChIP-seq中的Fastq文件与人类基因组的hg19序列进行比对,参数如下:-m1-t-best-q-S-l32-e80-n2。通过比较dexamethasone诱导的细胞GR ChIP-seq与乙醇处理的GR ChIP-seq,以及输入对照文库的STAT3 ChIP-seq,确定ChIP-seq峰。使用基于模型的CHIP-SEQ-2分析(MACS2)调用峰,P值截止值为1e-10,mfold参数限制在15和100之间。Bedtools Merge用于合并来自每个ChIP-seq实验的MACS2窄峰bed文件。在每个细胞系的每个ChIP-seq实验中,使用Bedtools overageBed来提取每个合并峰值下的读取数据。DESeq2版本1.20.0用于识别阅读深度在基底样细胞系和管腔细胞系之间有显著差异的峰(校正后P值0.05)。Pheatmap package version 1.0.10和Deeptools Version 3.1.0 computeMatrix和plotHeatmap函数用于创建ChIP-seq数据的热图。
RNA-seq
细胞系的RNA-seq已在前面描述过,这些数据可通过NCBI GEO登录 GSE58135公开获得。
单独或联合dexamethasone、STAT3 siRNA和SH4-54处理的MDA-MB-231和HCC70细胞株的RNA-seq结果如下:
STAT3 siRNA敲除采用GE Healthcare(L-003544-00-0005)ON-TARGET plus Human STAT3 siRNA kit(L-003544-00-0005)。此SMARTpool siRNA包含四个汇集的siRNA,每个siRNA针对STAT3 RNA序列的一个单独区域。ON-TARGETplus非靶向siRNA#1(D-001810-01-05)是一种非靶向对照。SiRNA SMARTpooll和非靶向siRNA靶序列如下:
ON-TARGETplus SMARTpool siRNA J-003544–07, STAT3:
GAGAUUGACCAGCAGUAUA
ON-TARGETplus SMARTpool siRNA J-003544–08, STAT3:
CAACAUGUCAUUUGCUGAA
ON-TARGETplus SMARTpool siRNA J-003544–09, STAT3:
CCAACAAUCCCAAGAAUGU
ON-TARGETplus SMARTpool siRNA J-003544–10, STAT3:
CAACAGAUUGCCUGCAUUG
ON-TARGETplus Non-targeting siRNA #1: UGGUUUACAUG-
UCGACUAA
siRNA转染实验在6孔板上进行,一式三份。根据说明使用脂质体RNAiMAX转染试剂(Thermo Fisher Science)。每孔含细胞,每孔加入250μLsiRNA-转染试剂混合物,最终浓度为25pmol siRNA,每孔7.5μL脂质体RNAiMAX试剂。
Dexamethasone处理采用6孔板,一式三份。用100nmol/L dexamethasone(D4902,Sigma Aldrich)或等体积100%分子生物级别乙醇载体对照(E7023,Sigma Aldrich)处理细胞4h。细胞用含10%β-巯基乙醇的RL缓冲液(Norgen Biotek)裂解。用动物组织RNA纯化试剂盒(Norgen Biotek)提取总RNA。
通过polyAselection (Dynabead mRNA纯化试剂盒,Invitgen)从250 ng总RNA中制备用于STAT3 siRNA和dexamethasone处理的RNA-seq文库,然后构建转座酶介导的非单链文库。文库在Illumina HiSeq 2000或HiSeq 2500测序仪上使用成对的50bp读数和6bp索引读数进行汇集和测序。Tophat v1.4.1用于将RNA-seq成对读数与GENCODE版本9进行比对。Cufflinks v1.3.0和BEDTools用于计算每个GENCODE转录本的原始数值。
用DESeq2版本1.20.0分别鉴定各细胞系在处理和对照条件下表达差异显著的基因。为了可视化具有不同表达的细胞系之间的基因差异表达,将对照样本中的基因表达除以平均值,并使用Pheatmap package version 1.0.10在R中的热图里显示归一化值。
对于GR和STAT3相互作用的研究,MDA-MB-231细胞被接种在6孔板中,在每孔4mL的培养基中约有30万个细胞,培养24小时。3个重复样本分别用1 μmol/L dexamethasone(DEX)、8 μmol/L SH4-54、不处理或dexamethasone+SH4-54联合处理24h。用350μL Qiagen RLT缓冲液和1%BME裂解细胞。RNA提取采用Norgen动物组织RNA纯化试剂盒。使用Illumina TruSeq Stranded RNA试剂盒和Ribo-Zero Gold制备RNA-seq文库,并在Illumina HiSeq2500上进行测序,作为50个循环的单端读取。使用带有参数-dta-p28-t-q-U的HISAT2版本2.1.0进行读数,并使用Samtools进行排序。使用FeatureCounts版本1.5.1从bam文件生成数据文件。
为了找出受GR和STAT3共同作用和协同调节的基因,我们使用了R中的线性模型(lm)函数。DESeq2归一化计算符合多变量模型,该模型包括GR活性项、STAT3活性项和交互作用项(GR:STAT3)。各基因的系数和P值用于识别受GR和STAT3的可加性和协同正、负调控的基因。协同作用被定义为交互作用项的的P值显著(0.05)。可加性被定义为相互作用项的P值非显著,以及同向系数的单独GR和STAT3项的P值显著。线性模型中的系数被用来确定GR和STAT3之间的相互作用导致了基因表达的增加还是减少。
来自STAT3 siRNA处理的RNA-seq数据可以通过NCBI GEO登录 GSE85579公开获得。
dexamethasone和SH4-54处理的RNA-seq数据可以通过NCBI GEO登录 GSE152201和GSE137535公开获得。
Kaplan-Meier分析
Kaplan-Meier绘图仪工具(参考http://kmplot.com;32),分析基因表达与无复发生存率的关系。Kaplan-Meier图是使用公开的基因表达数据生成的,这些数据来自有无复发的生存肿瘤患者。使用Kaplan-Meier绘图仪工具提供的现有的亚型分类来选择基底样病例和管腔病例进行分析。对GR、STAT3和438个由GR和STAT3上调的基底样基因进行了分析。所有的分析都采用了以下选择:每个基因只使用一个JetSet最佳探针,多基因使用所选探针的平均表达,选择无复发生存率进行分析,在随访阈值(60或120个月)对患者进行调查,排除有偏移的数据,并删除多余的样本。根据最显著的数值将患者分成两组(“自动选择最佳截断点”选项)。
细胞生长的实时成像
使用IncuCyte Zoom活细胞成像系统(Essen BioSciences)进行分析。将MDA-MB-231和MDA-MB-436细胞接种于96孔板中,每孔20,000个细胞。在进行滴定实验时,SH4-54的剂量范围为0~11 μmol/L,dexamethasone的剂量范围为0~2 μmol/L,联合处理时细胞分别加入8 μmol/L的SH4-54和1 μmol/L的dexamethasone。每隔2小时扫描一次平板,连续72小时,用IncuCyte ZOOM软件计算细胞联合度。通过将每个时间点的联合百分比减去第一个时间点的起始联合百分比来计算相对联合百分比。
患者源发性类器官样本培养试验
这项实验使用了两种源自患者的器官培养物,它们来自于原发的基底样TNBC肿瘤(HCI -001和HCI -002)。培养液为Advanced DMEM/F12、(Thermo Fisher 12634028)、5% FBS (Thermo Fisher 26140079)、1×Hepes (Thermo Fisher 15630080)、1×GlutaMAX Supplement (Thermo Fisher 35050061), 1μg/mL hydrocor-tisone (Sigma Aldrich H0888),10ng/mL hEGF (Sigma Aldrich E9644), 50μg/mL gentamicin (Genesee 25-533),10 μmol/L Y-27632 (Selleckchem S1049)。如前所述,在384孔板的每孔中,将有类器官组织分为50-100种接种在5%Matrigel(康宁354230)中。本实验以MDA-MB-231细胞株为阳性对照,在384孔板中以每孔500个细胞的速度培养。用SH4-54(0、1、2.1、3.2、4.3、5.4、6.5、7.6、8.8、9.9和11 μmol/L)和mifepristone(0、0.1、1.42、2.73、4.05、5.37、6.68和8 μmol/L)的所有可能组合处理细胞。72小时后,使用测量ATP的CellTiter Glo 3D(Promega)测量细胞存活率。应用Loewe加性分析计算协同性。
结果
基底样乳腺癌特异性调控区和候选转录因子的鉴定
我们假设,与管腔型乳腺癌相比,基底样型乳腺癌中特异性未甲基化的基因组调节区将富含参与控制基底特异性基因表达的TF结合位点。为了确定基底样乳腺癌的特异性调控区域,对28个乳腺癌细胞株系进行了RRBS,以测量整个基因组的DNA甲基化。这些细胞系包括18个先前被归类为基底样细胞的细胞系,以及10个先前通过PAM50亚型被归类为管腔的细胞系。这28个基底样细胞和管腔细胞系代表了TNBC内发现的异质性,包括以前被归类为间充质和LAR亚型的细胞系,以及具有BRCA1和BRCA2突变的细胞系(图1A)。在基因组中至少10倍覆盖率的479,746个CG位点中,3,748个CG位点在基底样细胞和管腔细胞系之间存在显著差异甲基化(线性回归Benjamini-Hochberg校正P
在腔性乳腺癌中,基因组的基因间隔区或启动子区域有1300个CG是显著未甲基化的。这些CG位点通过ENCODE Project发现在不同细胞系中对149个TF进行的ChIP-seq实验,TF结合位点的位置有相交部分。为了确定哪些TF在管腔乳腺癌细胞中特异性未甲基化的区域富集,将与这些CG相交的每个TF的结合位点数值除以ENCODE数据中每个TF的结合位点的数值(补充表S1)。序列特异性TF结合位点最高的是ER(6.9倍)、FOXA1(8.1倍)和GATA3(10.3倍)。这些结果证实了这种方法是合理的,因为ER、FOXA1和GATA3是已知的管腔基因表达程序的主要调节因子(图1A)。
在基底样乳腺癌中,基因组的基因间隔区或启动子区域有1793个CG是特异性去甲基化的。这些CG位点通过ENCODE Project发现在不同细胞系中对149个TF上进行的ChIP-seq实验,TF结合位点的位置相交的部分。这些区域中富集程度最高的特异性TF是JUN和FOS TF,它们通过异源二聚形成AP-1复合物(6.2倍)、STAT3(4.8倍)和GR(4.2倍;图1A)。这一结果使我们可以假设JUN、STAT3和GR参与调控基底样乳腺癌基因的表达。
为了进一步研究这一假设,我们试图使用初代人类肿瘤而不是细胞系,通过ATAC-seq直接研究染色质的可及性,而不是从缺乏DNA甲基化来推断它,并使用TF基序富集而不是ENCODE Project ChIP-seq实验,该实验不包括任何基底样乳腺癌细胞系。我们分析了59个原发性乳腺肿瘤的ATAC-seq数据,其中包括15个基底样肿瘤和44个管腔肿瘤,这些肿瘤按照PAM50亚型分类。与腔型肿瘤相比,基底样肿瘤中有110,156个区域特异性地呈现开放染色质(DESeq2校正P
同时抑制GR和STAT3可协同抑制细胞生长
通过得到的数据我们观察到了GR和STAT3协同调节了大量与细胞增殖和不良预后相关的基因表达信 中的基因,这促使了我们开始研究调节GR和STAT3的药物是否会影响细胞生长。为了初评GR和STAT3在细胞生长中的独立作用,分别用不同剂量的dexamethasone和STAT3抑制剂SH4-54处理MDA-MB-231细胞,并在Incucyte Zoom活细胞成像仪上监测72小时。随着STAT3抑制剂SH4-54剂量的增加,STAT3活性降低会导致细胞生长减少,并对剂量呈现出依赖性(图7A)。通过减少dexamethasone的量来降低GR活性会导致细胞生长以剂量依赖的形式减少(图7B)。这些结果与以前 道的分别抑制GR和STAT3会分别抑制基底样乳腺癌增殖的 道是一致的。
为了评估同时调节这两种TF是否会导致生长差异,我们用dexamethasone和SH4-54的不同组合处理了MDA-MB-231和MDA-MB-468细胞。同时用dexamethasone抑制GR和8μmol/L的SH4-54抑制STAT3,细胞生长最慢(图7C和D)。相反,用1μmol/L dexamethasone激活GR再结合STAT3的内源活性,细胞生长最快(图7C和D)。值得注意的是,当GR或STAT3单独活跃时,会导致中等的增长率。这一结果支持了GR和STAT3协同促进细胞生长的假设,同时抑制GR和STAT3会导致比单独抑制使细胞更慢的生长。
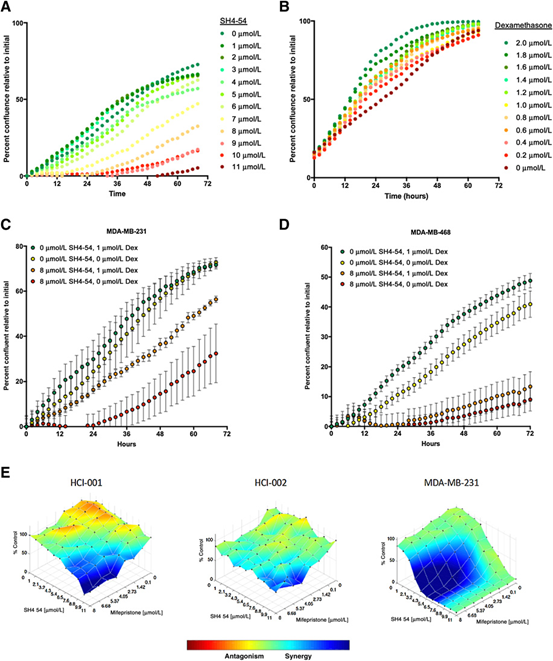

图7抑制GR和STAT3导致基底样细胞系和患者源发性类器官样本细胞的生长减少。随着STAT3抑制剂SH4-54剂量的增加,MDA-MB-231细胞的生长速度呈剂量依赖性下降。降低dexamethasone剂量对MDA-MB-231细胞生长有抑制作用,且呈剂量依赖性。与单独抑制相比,同时抑制GR和STAT3对MDA-MB-231?和MDA-MB-468(D)细胞的生长均有抑制作用。患者源发性类器官样本细胞培养物(HCI-001和HCI-002)和MDA-MB-231细胞系同时用增加剂量的STAT3抑制剂SH4-54和GR抑制剂mifepristone处理72h后,细胞生长(蓝色)协同下降。
讨论
本研究的目的是确定哪些TF驱动碱基样基因表达程序。对DNA甲基化、染色质可及性、TF结合和基因表达的全基因组分析表明,GR和STAT3会丰富地结合在基底样乳腺癌中特异开放的基因组调节区。这些转录因子与相同的调控区域结合,协同调节数百个基底样表达信 中的基因,包括许多其他转录因子。GR和STAT3协同上调的基因是基底样型乳腺癌生长的主要信 转导通路(EGFR和TGFb),与基底样型乳腺癌的侵袭特征(增殖、干性和上皮向间质转化)相关,并与患者较短的无复发生存期有关。此外,同时抑制GR和STAT3会导致多种细胞系和患者源发性类器官样本细胞培养中细胞生长的协同减少。总之,这些数据表明,同时抑制GR和STAT3可以减少基底样基因表达信 的表达,并创造出一种可用于治疗的合成致死性。
尽管GR和STAT3分别参与了基底样乳腺癌基因表达的调控,但它们的协同作用尚未被 道。本研究清晰地阐明了,了解这种相互作用对于研究它们在驱动基底样基因表达和表型方面的作用以及研究出更有效的治疗方案至关重要。单独抑制GR是无效的,基底样细胞系在培养基中没有糖皮质激素的情况下增殖并表达基底样基因信 的某些成分。单独抑制STAT3并不是完全有效的;先前的研究表明,SH4-54在培养中会使基底样细胞系致死,但不足以在体内完全阻止肿瘤的生长,因为体内会自然存在糖皮质激素。这些结果表明,单独抑制这两种转录因子中的任何一种都不足以在体内减少基底样癌细胞的增殖基因表达,但本研究表明同时抑制GR和STAT3可能是一种新的有效的治疗策略。
有大量之前的文献可以支持我们的结论,即STAT3是乳腺癌细胞生长、侵袭和存活的关键因素,包括我们之前的研究表明,STAT3结合调节促进侵袭的基因。由于JAK/STAT3途径抑制剂的致癌作用已知,目前已有多项临床试验测试了JAK/STAT3途径抑制剂在乳腺和其他实体肿瘤中的作用,如图3所示。这些试验,以及其他直接针对STAT3的药物的不断开发,为未来的研究提供了很大机会,以确定与GR抑制剂联合治疗是否可以提高疗效,或者联合治疗是否可以以更低、更好的耐受性剂量提供类似的治疗效果。
研究GR在基底细胞样乳腺癌中的作用具有重要的临床意义。GR在癌症中的作用是疾病和亚型特异性的。GR的表达与管腔性乳腺癌的良好预后相关(图2D),糖皮质激素通常用于治疗血液系统恶性肿瘤,因为它们诱导淋巴细胞凋亡。此外,强力合成糖皮质激素经常被用于接受过化疗的乳腺癌患者,以防止恶心、食欲不振和罕见的严重免疫反应等副作用。我们的初步数据支持其他几项研究结果,即糖皮质激素诱导GR可以促进TNBC的肿瘤生长和治疗耐药性。具体地说,在我们的研究中,糖皮质激素诱导GR导致基底样乳腺癌细胞系和患者源发性类器官样本的细胞生长增加是显而易见的。它还导致受STAT3协同调控的基因表达增加,STAT3是已知的癌症生长和侵袭的促进剂。最近,临床试验已经开始测试直接用mifepristone抑制GR还是通过抑制其伴侣HSP90来提高ER阴性乳腺癌的化疗疗效。虽然糖皮质激素的使用被认为是提高化疗耐受性的必要手段,但mifepristone联合nabpaclitaxel抑制糖皮质激素受体的首次试验表明,使用filgrastim生长因子可以控制主要的毒性,即嗜中性白血球减少症。这项研究发现,标准剂量的nabpaclitaxel与有效剂量的mifepristone联合使用是安全和耐受的,该方案的II期试验已经启动。补充图中提供了GR和Hsp90抑制剂的目前试验列表S4。我们的研究结果支持这一研究方向,并显示同时抑制GR和STAT3的联合治疗在治疗基底细胞样TNBC方面可能更有效。
原文解读:点击查看全文
声明:本站部分文章及图片源自用户投稿,如本站任何资料有侵权请您尽早请联系jinwei@zod.com.cn进行处理,非常感谢!