对两处温泉的泥浆沉积物样本的不同水平物种分类(图3-图8)展示出了两个群落的病毒多样性。研究发现两个温泉都富含dsDNA。并且两个温泉的泥浆沉积物样本中的宏病毒组序列的优势目包括有尾噬菌体目(Caudovirales)、孢疹病毒目(Herpesvirales)和逆转录病毒目(Ortervirales)。此外,研究在NYS温泉样本中发现了属于未分类的巨型DNA病毒的序列;仅在NYS样本中发现具有ds(RT-DNA)的巴德纳病毒(Badnavirus)的序列,并仅在OYS样品中发现具有ss(-)RNA的风疹病毒(Rubulavirus)的序列。
从属水平上看,两温泉样本共有的病毒包括:Pandoravirus(潘多拉病毒)、Mimivirus(拟菌病毒)、Nonanavirus、Alphabaculovirus(α杆状病毒)、Prasinovirus(普拉斯诺病毒)、Fromanvirus、Varicellovirus(水痘病毒)、Phicbkvirus、Marseillevirus(马赛病毒)、Timquatrovirus,它们构成了病毒圈的主要种群。
从种水平上看,两温泉样本共有的病毒包括:BeAn 58,058 virus、Human endogenous retrovirus K(人内源性逆转录病毒K)、Moumouvirus、Pandoravirus salinus、Pandoravirus macleodensis、Megavirus chiliensis、Pandoravirus dulcis、Noumeavirus、Acanthamoeba polyphaga mimivirus、Pandoravirus neocaledonia。
根据病毒功能基因多样性分析,发现NYS温泉泥沉积物以细菌噬菌体(六个主要物种)为主导。其中主要的10个物种分别是:肺炎球菌噬菌体Dp-1,棘阿米巴多噬拟态病毒(APMV),葡萄球菌噬菌体L54a,鸡痘病毒(FPV),双斑侧沟茧蜂病毒(MdBV),埃希氏菌噬菌体Mu,芽孢杆菌噬菌体phi105,棉铃虫颗粒病毒(HaGV),埃希氏菌噬菌体lambda和大肠杆菌噬菌体N15。相似地,在OYS温泉泥沉积物中,也是细菌噬菌体(八个主要物种)占主导地位。在OYS温泉的宏病毒组功能序列中发现了一种主要的古细菌噬菌体ATV。OYS样本中主要的10个物种分别是:芽孢杆菌噬菌体SPbeta、埃希氏菌噬菌体P1、肺炎球菌噬菌体Dp-1、棘阿米巴多噬拟态病毒(APMV)、埃希氏菌噬菌体λ、酸菌双尾病毒(ATV)、沙门氏菌噬菌体P22、艾克沙门氏菌病毒、大肠埃希氏菌噬菌体λ和大肠埃希氏菌噬菌体Mu。
研究使用基于四核核苷酸频率的平均距离矩阵,计算了两个样本共有的宏病毒组圆形 络模型。每个病毒的序列作为一个单节点,节点大小取决于连接到节点的边权重, 络的边显示出病毒基因组及其核苷酸频率丰度的相关性,边的权重与节点之间的频率相关。病毒组的 络分析图展示出了它们对真核生物,细菌和古细菌共同宿主的概率相互依赖性。
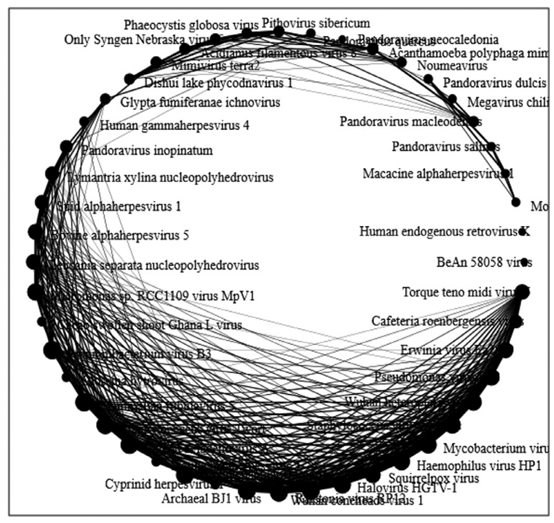

图9:基于四核苷酸频率的平均距离矩阵的宏病毒组 络分析图
结论:
该研究利用锡金两个温泉含硫泥浆沉积物的宏基因组数据,分析了该生态系统中宏病毒组的组成及其功能多样性。研究通过Alpha多样性和beta多样性展示了两个群落的多样性特征,描述了两个取样地点的群落结构、共有的病毒种类和两地特有的物种。研究根据病毒功能基因多样性,发现细菌噬菌体是两处温泉泥沉积物样本中最主要的成分。此外,根据四核苷酸频率的平均距离矩阵构建了共有病毒 络分析图,展示出它们共同宿主的概率相互依赖性。这些发现可以作为可培养病毒学研究动态的依据。
参考文献:
Das, S., Kumari, A., Sherpa, M. T., Najar, I. N., & Thakur, N. (2020). Metavirome and its functional diversity analysis through microbiome study of the Sikkim Himalayan hot spring solfataric mud sediments. Current Research in Microbial Sciences. doi:10.1016/j.crmicr.2020.05.002
文中病毒组研究方法:
- 序列组装采用的是metaSPADES assembler (Nurk et al., 2017)。该软件是目前宏基因组领域组装指标较好的软件,尤其在株水平组装优势明显。
- 组装结果分箱采用的工具是MaxBin 2.0 (Wu et al., 2016)。Maxbin考虑每个contig的序列覆盖度和四碱基频率,以记录每个bin的marker基因数量。
- 分类采用的是Kraken2 (Wood et al., 2019),参考数据库包含所有的Ref-Seq细菌、古细菌、真菌和病毒基因组。Bracken (Lu et al., 2017)用来评估从种水平到门水平的不同微生物丰度。
- 采用Prodigal (Hyatt et al., 2010)对蛋白质编码基因(ORFs)进行预测,采用ARAGORN(Laslett and Canback, 2004)对tRNA基因进行预测,并且采用rRNAFinder(Wheeler and Eddy, 2013)对核糖体RNA基因进行鉴定和分类。
- 计算多样性指数、稀疏曲线和 络分析采用的软件是PAST (Najar et al., 2020)。
技术文章链接:点击了解详情
声明:本站部分文章及图片源自用户投稿,如本站任何资料有侵权请您尽早请联系jinwei@zod.com.cn进行处理,非常感谢!